
English follows Japanese (Jump to English version)
機械学習の有用性に疑問の余地がなくなった現在、有機合成分野でのデータ科学の最重要課題のひとつとしてデータ基盤の整備、すなわちその中核となる反応設計に直結するデータベースの構築があげられます。本ブログではとくに分子触媒を対象にしていますが、これまでも分子触媒設計のためのデータベースが提案されてきました。それらの機械学習を用いた応用研究も少しずつ報告されるようになっています。そのなかのひとつとして、本記事では、分子触媒設計のための新たなデータ基盤の構築に向けた取り組みと、その活用方法を提案します。
カギとなるのが機械学習+遷移状態計算による触媒設計です。遷移状態計算で収集した「触媒活性」と「遷移状態構造」の機械学習により触媒設計が可能となることを前回述べました。今回は遷移状態計算で収集した「触媒活性」と「遷移状態構造」からなるデータベースを用いた以下のようなデータ利活用法および触媒設計プロセスを提案します。
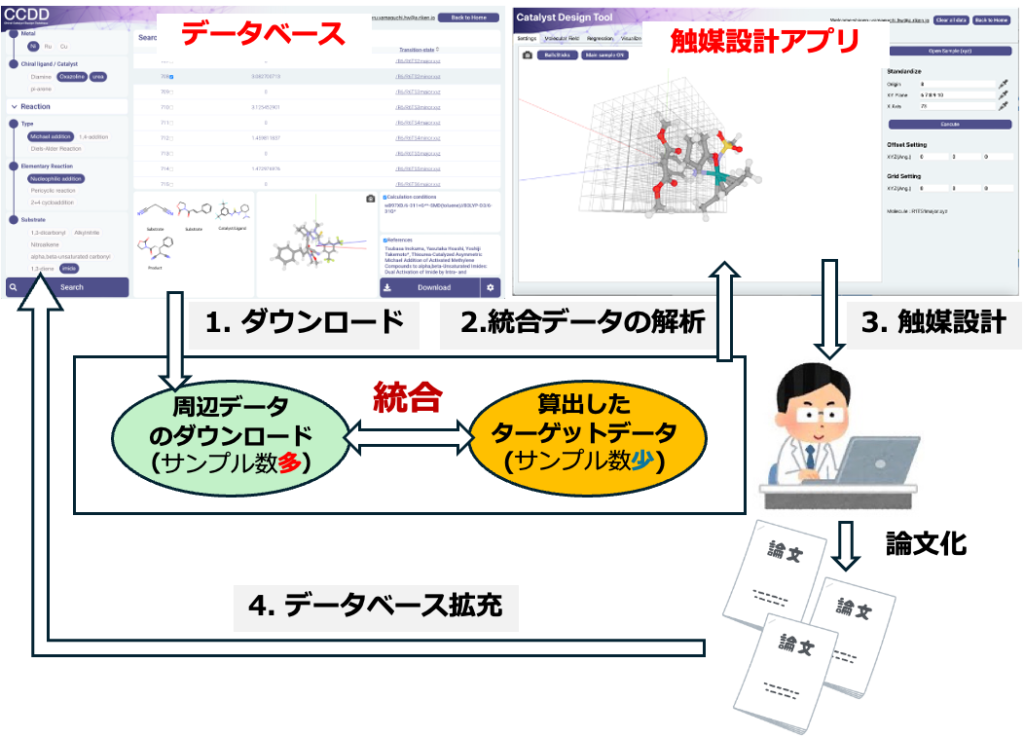
1. ダウンロード:データベースから、開発中の反応と類似したデータ(触媒活性+遷移状態構造)をダウンロード
2. 統合データの解析:ダウンロードしたデータと、手元にある開発中の反応の少数データを統合しデータ解析
3. 触媒設計:データ解析をもとに触媒設計
4. データベース拡充:設計した触媒に関するデータを論文とともに公開このプロセスが実現すれば、反応発見後の触媒最適化の労力が格段に減ります。また触媒設計を行った際に創出したデータをデータベースに格納することで、自然と機械学習用データが拡充されます。この流れを図示すると、機械学習用データの「創出」、創出したデータのデータベース収納による「統合」、データの「利活用」というデータ循環のサイクルが形成されていることがわかります(下図左)。
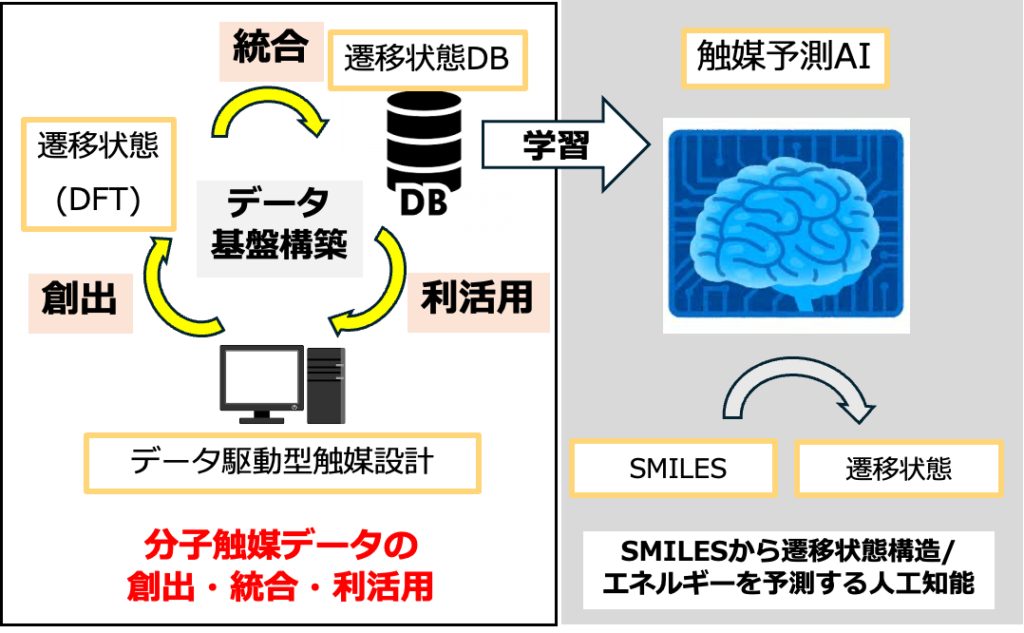
本サイクルを継続的に循環させ、良質なデータが大量に出そろえば、化合物を表す文字列(SMILES)から遷移状態構造およびそれにともなう遷移状態エネルギー(触媒活性)を予測する人工知能が構築できます(上図右)。このプロセスは、創薬研究などで高い有用性を持つために、タンパク質の単結晶X線構造が世界中で継続的に「創出」され、それらがPDB (Protein Data Bank)として「統合」され、研究に「利活用」されるというAlphaFold2の構築に寄与したデータ循環と類似するものではないかと考えています。
この循環を回し出すために必要な、上図で示したデータベースからダウンロードしたデータと、開発中の反応データとの「統合」に基づく触媒設計に関して、計算機上ですが不斉触媒反応を対象に概念実証に成功しています(J. Org. Chem. 2025, 90, 17518.)。
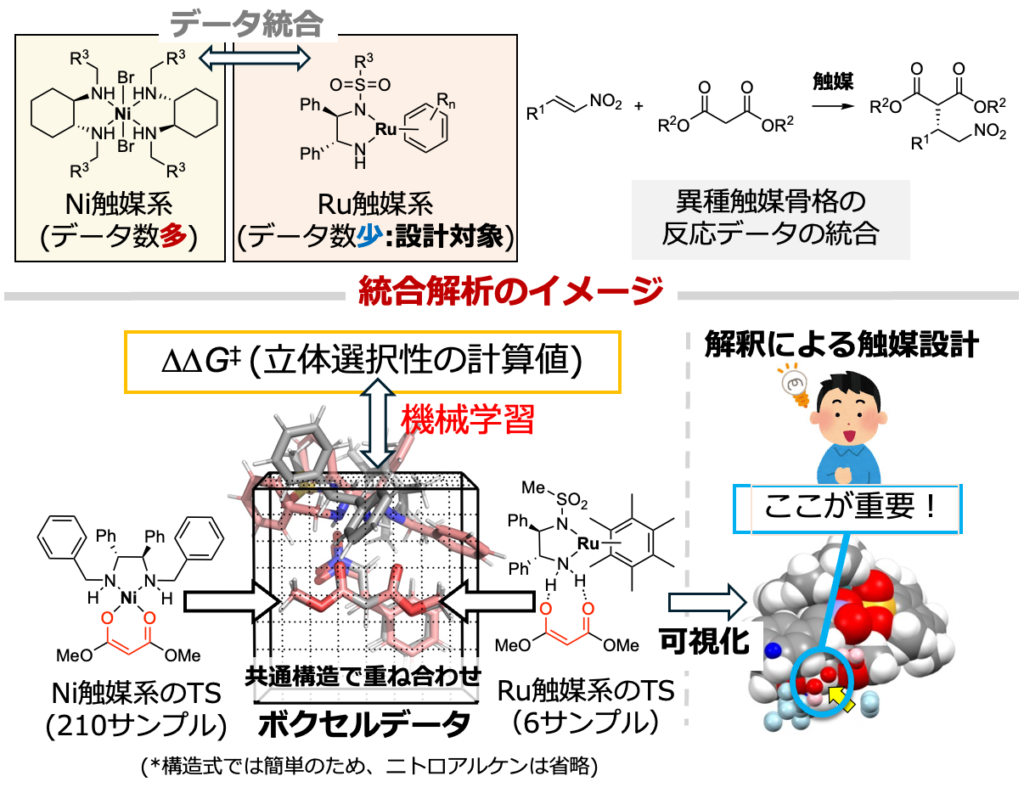
図に示すように、Ni触媒(210サンプル)とRu触媒(6サンプル)による不斉マイケル付加を解析対象とし、異種触媒骨格を用いた反応の統合解析を行いました。赤くハイライトした共通の反応部位で遷移状態構造を重ね合わせし算出したボクセルデータ(2次元画像データであるピクセルデータの3次元版)と、∆∆G‡値(立体選択性の計算値)とを機械学習で相関づけすることで、遷移状態のどこが重要かを可視化、その情報をもとに、立体選択性の計算値が向上するRu触媒の設計に成功しています。多数の反応データ(210サンプル:Ni触媒による不斉マイケル付加)と少数の反応データ(6サンプル:Ru触媒による不斉マイケル付加)から、少数反応データ側の触媒の最適化を行っています(Ru触媒のデータの解析のみでは、触媒設計のための情報を抽出することはできません)。既知反応を対象とした解析例ですが、上記の結果はデータベースからダウンロードしてきた類似の反応データとの統合解析により、手元にある開発中の反応の触媒設計が可能であることを示しています。
論文(J. Org. Chem. 2025, 90, 17518.)では、上記Ni触媒/Ru触媒系に加え、金属触媒系と有機触媒系の統合解析や、マイケル付加とディールス・アルダー反応の統合解析など、計7種の統合データセットの解析に基づく計算機上での触媒設計に成功しています。
有機合成のデータ科学の潮流の一つである、Hammett則から発展してきた自由エネルギー関係に基づく回帰分析では分子骨格を揃え、置換基をスクリーニングすることで良質なデータを収集・解析することが常識でした。分子骨格を揃えて解析を行うため、この分野で代表的なHammett σやTaft値などの記述子は、置換基毎に定められた値を取ります。つまり分子骨格が異なっていても同じ置換基を使っている場合は、記述子の値が同じになります。その結果、異なる分子骨格上に置かれた同一置換基の環境差を反映することが難しく、異種反応データの統合解析は困難でした。今回、反応中心で重ね合わせた遷移状態構造をボクセルデータに変換することにより、分子骨格の違いに基づく反応点周りの環境の変化を比較可能としました。異種反応データの統合解析が解釈性の高い形で可能となり、触媒設計につながっています。データ収集にはDFT計算を用いていますが、実験と異なり、データ算出条件を揃えやすいために、統合解析に適した均質性の高いデータを創出できることも重要な点です。
本触媒設計プロセスが一般化すれば、上述のように、分子触媒データの創出・統合・利活用というデータ循環のサイクルが形成され、PDBのようなデータ基盤の構築につながる可能性があります。そこで、このデータ循環のサイクルを加速するために、上述の分子ボクセルデータ解析のための触媒設計アプリおよび機械学習用データベースからなるデータ駆動型触媒設計システムを構築し、学術研究機関向けに公開しています(https://mcds.riken.jp)。
以上、今回は、前回述べた機械学習+遷移状態計算による触媒設計の発展型である異種反応のデータ統合に基づく触媒設計を基盤とした、分子触媒データの創出・統合・利活用のデータ循環サイクル、ひいては有機合成分野のデータ基盤の構築プロセスを提案しました。
一般に、NMRに代表される分析手法の出現は、有機分子に基づく現象の理解を深化させ、分野そのものの発展につながります。不斉触媒反応やクロスカップリングといった分子触媒の枠組みの確立は、NMRなどの分析手法の進展に大きく支えられてきたと考えられます。ここで、機械学習を用いれば、遷移状態計算の結果を触媒設計が可能なレベルで解釈できます。この観点から、機械学習もまた、有機合成における分析手法のひとつと位置づけることができます。さらに、上述のように良質なデータが蓄積されれば、分子を表す文字列(SMILES)から遷移状態構造およびそのエネルギーを予測する人工知能の構築も現実的な目標になると考えられます。
NMRが1960年代に有機合成分野に普及される準備が整ったのち、1970年代以降は不斉触媒・クロスカップリング等、新しい分子触媒の枠組みが次々に確立されていった非常にワクワクする時代だったのではと想像します。上記のように、分析手法として(また予測手法としても)機械学習が有機合成分野に浸透しはじめています。機械学習を使えば、これまで不可能だと思われていたような複雑な触媒反応の制御が実現できるかもしれません。たとえば、有機合成の難題のひとつといわれている複雑な反応(立体分岐型不斉合成)を、分子のボクセルデータの解析に基づく触媒設計により制御できることを見出しています(Cell Rep. Phys. Sci. 2021, 2, 100697. : プレスリリース)。また、高分子誌2002年1月号の「20年後の高分子」という特集(https://doi.org/10.1295/kobunshi.51.18)の中に以下のような課題が掲示されています。
「面選択性(立体構造)、反応選択性(分子量)、基質選択性 (モノマー適合性と連鎖配列)などを厳密にしかも同時に規制しうる 「精密重合」は、一般的指針さえ未確立である。」
分子のボクセルデータに基づく機械学習を使うと、この重合触媒における課題のひとつである、複数のreaction outcomesの制御をある程度できることがわかっています(Catal. Sci. Technol. 2024, 14, 2434.)。こうした実際の研究における手応えから、今後、機械学習を活用して、これまで有機合成のチャレンジとされてきた数々の課題が解決できる可能性は低くないと思っています。あとから振り返った際に、有機合成の転換点ともいえるあの頃に研究に携われたのは幸運だったと思えるような、非常にエキサイティングな時代にいるのかもしれません。
筆者自身は、上記分子触媒データの創出・統合・利活用に関して、出願特許をもとに理研発スタートアップを立ち上げています(プレスリリース)。
また、機械学習用データベースを活用し、反応データを統合しながら触媒設計を行う研究の枠組みを「統合触媒科学」と位置づけ、現在、この考え方に基づく共同研究を進めています。その研究会を2026年3月16日、日本化学会年会前日に船橋で開催します(統合触媒科学研究会)。本研究会では、触媒反応の統合解析を通じて、機械学習が有機合成に浸透しつつある現在進行形の変化を議論します。加えて、触媒設計システムのチュートリアルも行う予定であり、参加者自身がこの新しい枠組みに触れ、研究者としてその変化に直接関わる可能性を提供する機会になればと考えています。ご興味ある方、参加希望の方は以下のポスターをご覧ください。

ここから英語版
Toward a PDB-like database for organic synthesis
As the usefulness of machine learning has become increasingly unquestionable, one of the most important challenges in data science for organic synthesis is the development of data infrastructure, namely, building databases that directly support reaction and catalyst design. Several databases for molecular catalyst design, which is the focus of this blog, have been proposed, and applied studies leveraging machine learning have been reported in recent years. This article presents an approach toward building a new data infrastructure for molecular catalyst design and proposes practical ways to utilize it.
The key concept is catalyst design driven by machine learning combined with transition-state (TS) calculations. The previous post discussed how machine learning models trained on TS-calculated data, namely catalytic activity and transition-state structures, can enable rational catalyst design. Building on this idea, this article proposes a workflow for data utilization and catalyst design based on a database composed of TS-derived catalytic activity and TS structural information, as shown below.
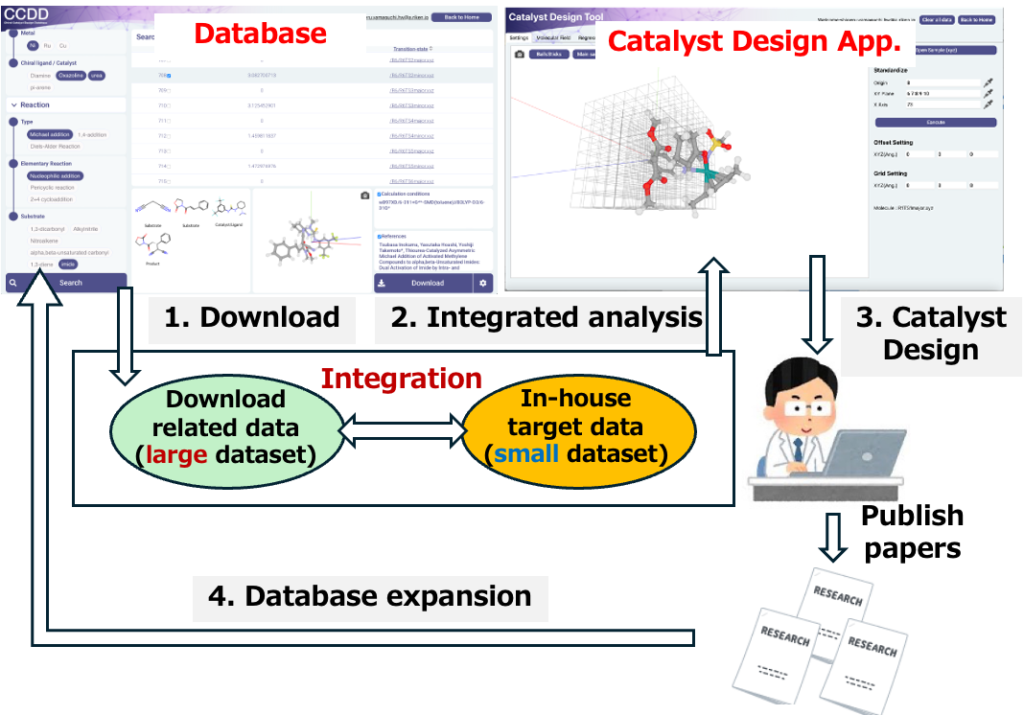
1. Download: Retrieve data similar to the reaction under development from the database (catalytic activity + transition-state structures).
2. Integrated analysis: Combine the downloaded data with the small amount of in-house data for the reaction under development, and perform data analysis.
3. Catalyst design: Design catalysts based on the results of the analysis.
4. Database expansion: Publish a paper along with the newly generated catalyst data, thereby expanding the database.If this workflow can be implemented, the effort required for catalyst optimization after reaction discovery can be dramatically reduced. Moreover, by depositing the data generated through catalyst design back into the database, machine-learning-ready data will naturally continue to accumulate. As illustrated in the figure (left), this creates a data-driven cycle consisting of data generation, integration through database deposition, and reuse of the data, enabling continuous and sustainable growth of the dataset.
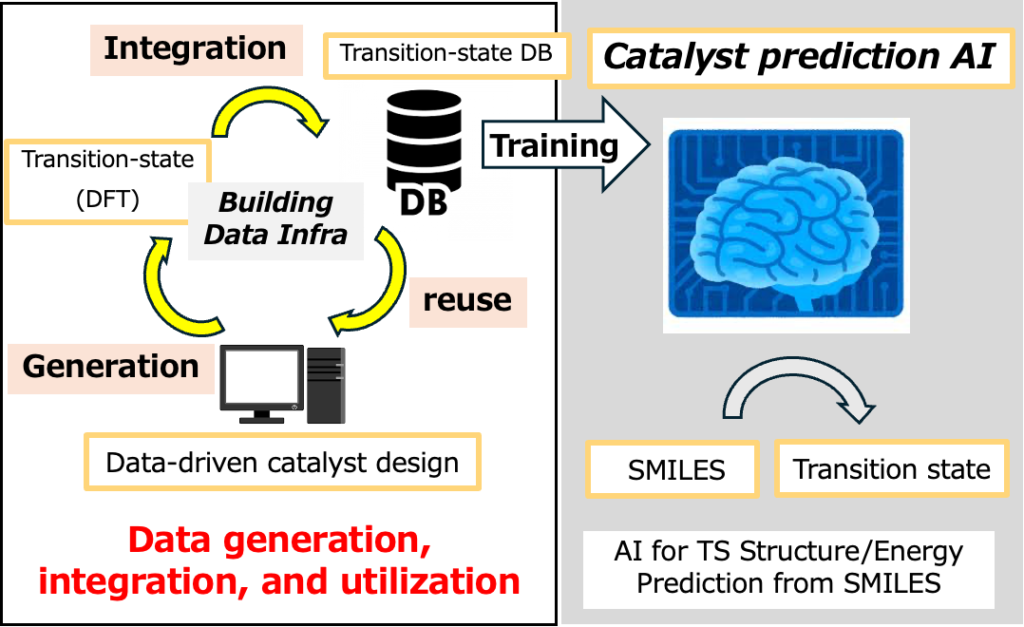
If this cycle is continuously repeated and a large amount of high-quality data becomes available, it will become possible to build an AI model that predicts TS structures and the corresponding TS energies (i.e., catalytic activity) directly from molecular representations such as SMILES (Figure, right). This concept may be analogous to the data ecosystem that supported the development of AlphaFold2, where protein single-crystal X-ray structures have been continuously generated worldwide, integrated into the Protein Data Bank (PDB), and broadly utilized in research due to their high value in drug discovery and related fields.
As a proof of concept for initiating such a cycle, this approach has been demonstrated computationally using asymmetric catalytic reactions. Specifically, catalyst design can be achieved through integrated analysis of relevant data downloaded from a database together with a small dataset constructed to represent a reaction under development (J. Org. Chem. 2025, 90, 17518.).
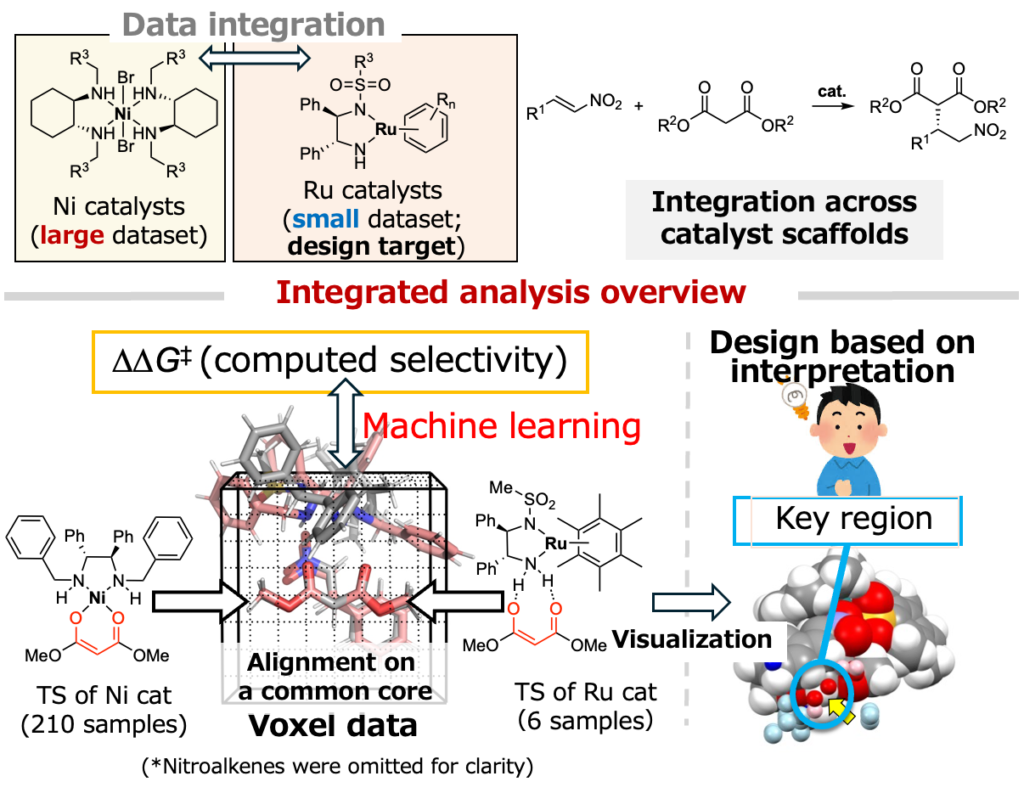
As shown in the above figure, an integrated analysis across different catalyst scaffolds was performed for asymmetric Michael addition reactions catalyzed by Ni catalysts (210 samples) and Ru catalysts (6 samples). TS structures were aligned at a common reactive site (highlighted in red) and converted into voxel descriptors (a three-dimensional analogue of pixel-based image data). By correlating these voxel descriptors with computed ∆∆G‡ values (i.e., computed stereoselectivities) using machine learning, the key regions within the TS structures that govern stereoselectivity could be visualized. Based on this information, Ru catalysts were successfully designed to improve the computed stereoselectivity.
In this approach, a large dataset (210 samples, Ni-catalyzed reactions) and a small dataset (6 samples, Ru-catalyzed reactions) were combined to optimize catalyst design for the small-data reaction system. Notably, design-relevant information cannot be extracted from the Ru dataset alone due to the limited number of samples. Although this example is based on known reactions, it demonstrates that catalyst design for a reaction under development can, in principle, be enabled through integrated analysis with similar reaction data downloaded from a database.
In the published study (J. Org. Chem. 2025, 90, 17518.), this concept was further validated across a total of seven integrated datasets, including integration between transition-metal catalysis and organocatalysis, as well as between different reaction types such as Michael additions and Diels–Alder reactions, leading to successful in silico catalyst design.
One of the long-standing approaches in data science for organic synthesis is regression analysis based on free-energy relationships, originating from the Hammett equation. In this framework, it has been common practice to collect and analyze high-quality data by keeping the molecular scaffold fixed and screening substituents. Because such analyses are performed on a consistent scaffold, widely used descriptors in this field—such as Hammett σ constants and Taft parameters—are defined primarily for each substituent. In other words, even when the scaffold changes, the descriptor value remains the same as long as the substituent is identical. As a result, it is difficult to capture differences in the local environment of the same substituent placed on different scaffolds, making integrated analysis across diverse reaction datasets challenging. In the present approach, transition-state (TS) structures aligned at the reaction center are converted into voxel descriptors, enabling direct comparison of structural environments around the reactive site even across diverse molecular scaffolds. This makes it possible to perform interpretable integrated analysis of diverse reaction data and to translate the results into catalyst design. Importantly, the dataset is generated using DFT calculations, which—unlike experimental measurements—allow computational conditions to be standardized more easily, resulting in highly homogeneous data well suited for integrated analysis.
If this catalyst design workflow can be generalized, it may establish a self-sustaining cycle of data generation, integration, and utilization for molecular catalyst research, ultimately leading to the development of a PDB-like data infrastructure for organic synthesis. To accelerate this cycle, a data-driven catalyst design platform has been developed and made available to academic research institutions, consisting of a catalyst design application for voxel-based analysis and a machine-learning-ready database (https://mcds.riken.jp).
In summary, this article has proposed a pathway toward building data infrastructure for organic synthesis through a data-driven cycle of generating, integrating, and utilizing molecular catalyst data, based on catalyst design enabled by integrated analysis of diverse reaction datasets—an extension of the machine-learning + transition-state calculation framework discussed in the previous post.
Building on this proposal, a research framework in which a machine-learning-ready database is utilized to integrate reaction data for catalyst design is referred to here as “Integrated Catalysis Science.” Collaborative research based on this concept is currently underway.
